Protein and Ligand Modeling
SPONGE 1.4 tutorial.This page was translated by GPT-5.5 AI.
Protein and Ligand Modeling
Last updated
2023/05/26
1. Introduction
This tutorial uses protein structure 4B1Y as an example to introduce how to build a model containing protein, ligands, and ions with Xponge.
Xponge version used: 1.3.4b3
CudaSPONGE version used: 1.3
2. Obtaining the Original Files and Basic Text Processing
Go to the 4B1Y page in the RCSB Protein Data Bank, then download the corresponding PDB file and the mol2 files for the small-molecule ligands needed in this tutorial, as shown below.
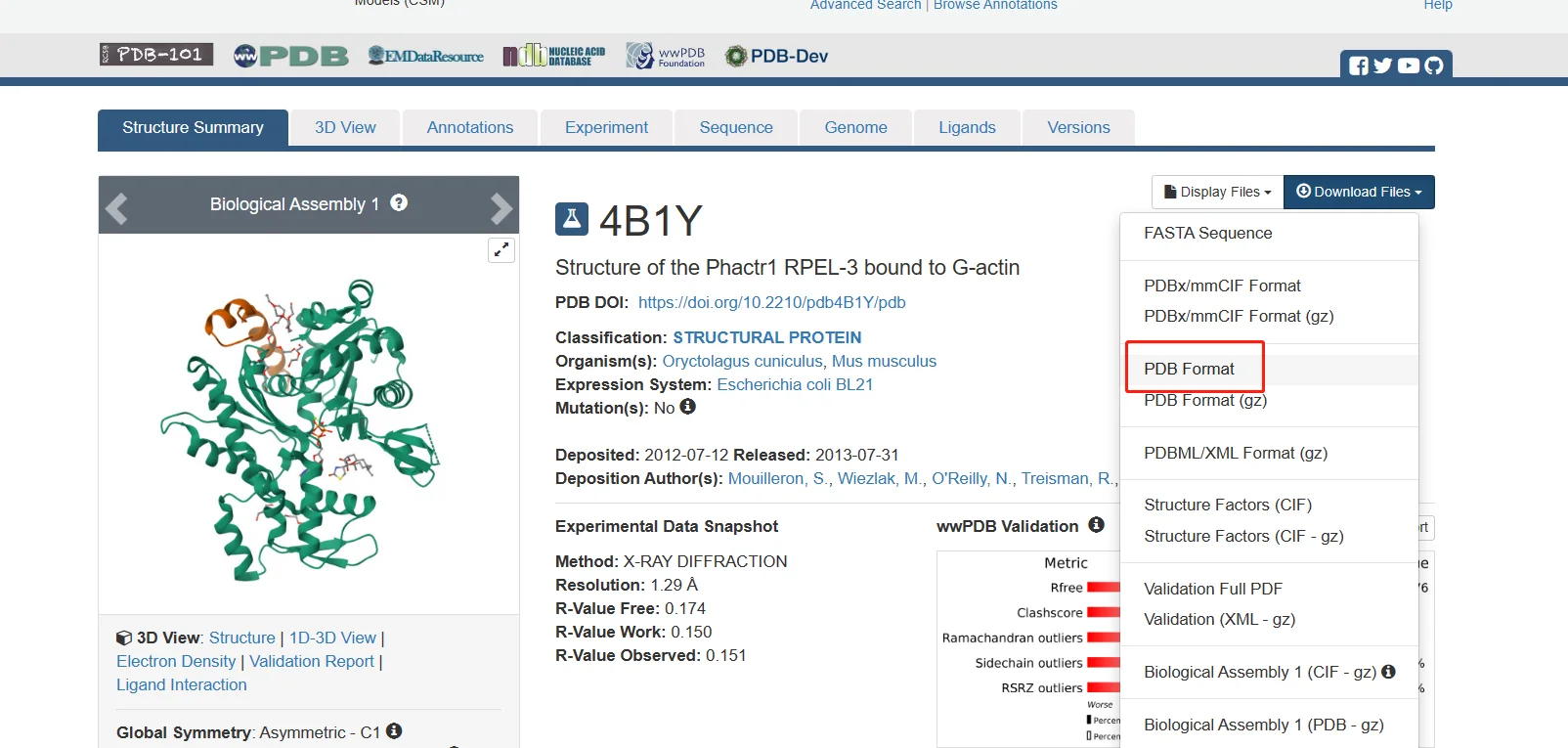
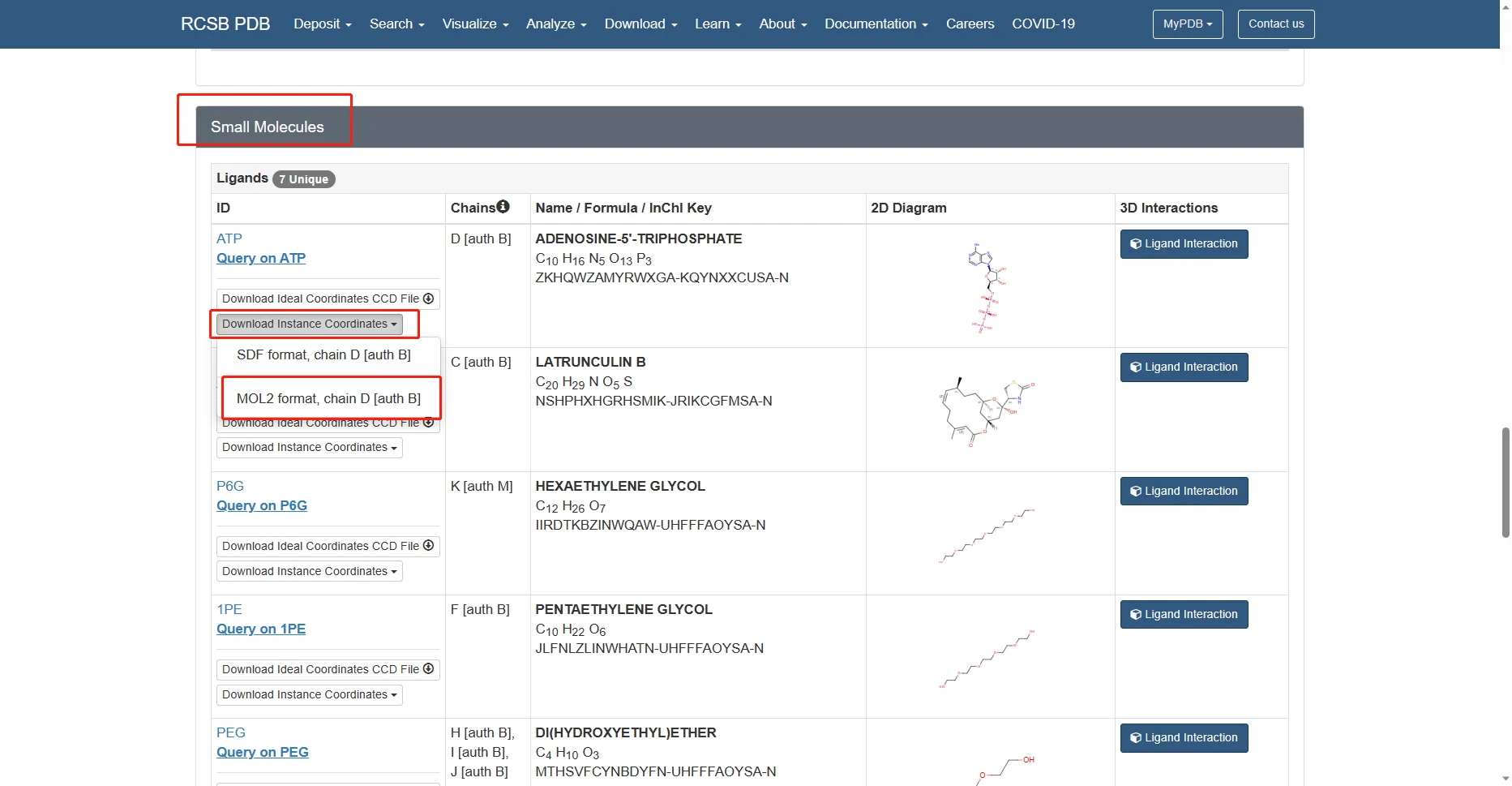
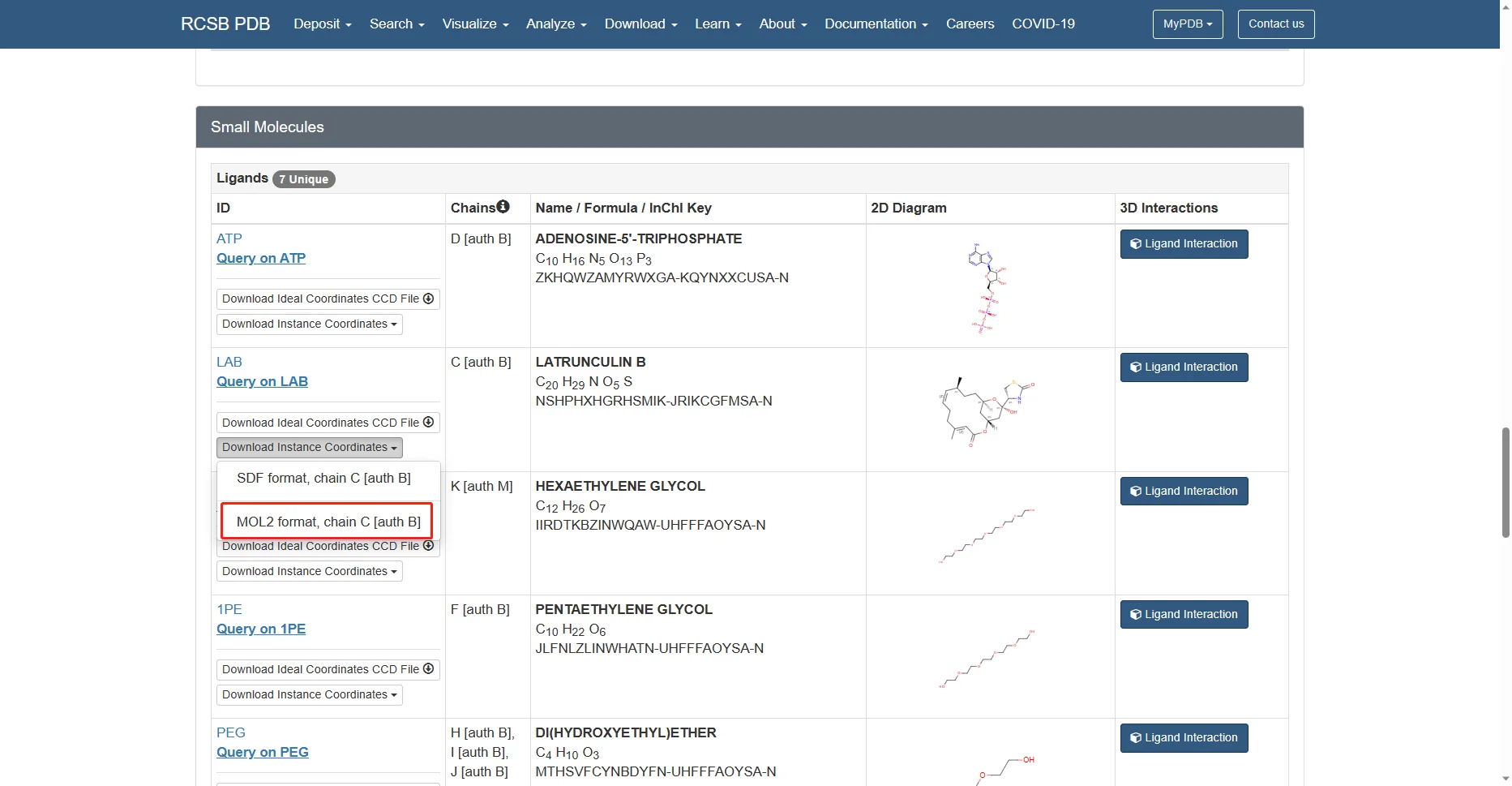
The remaining alcohol molecules and water molecules are small molecules from the protein crystallization process and are not needed, so they are not downloaded.
Use openbabel to add hydrogens to the mol2 files and set the pH to 7. After openbabel adds hydrogens, it changes the atom names. Use Xponge name2name to change the atom names back to those in the PDB file.
obabel 4b1y_D_ATP.mol2 -O ATP.mol2 -p 7
Xponge name2name -tformat pdb -tfile 4b1y.pdb -tres ATP -fformat mol2 -ffile ATP.mol2 -oformat mol2 -ofile ATP.mol2
obabel 4b1y_C_LAB.mol2 -O LAB.mol2 -p 7
Xponge name2name -tformat pdb -tfile 4b1y.pdb -tres LAB -fformat mol2 -ffile LAB.mol2 -oformat mol2 -ofile LAB.mol2
Although the PDB file also contains structural information for the small molecules, the information in the PDB file is incomplete: it does not contain bond order or hybridization information. When tools such as openbabel are used for conversion, errors may occur for some molecular structures. Therefore, it is best to download the mol2 files directly from the source website.
Open the downloaded PDB file and inspect the header. The REMARK 465 section shows that some residues are missing from this PDB file, so the SEQRES section needs to be kept in order to complete the missing residues.
REMARK 465
REMARK 465 MISSING RESIDUES
REMARK 465 THE FOLLOWING RESIDUES WERE NOT LOCATED IN THE
REMARK 465 EXPERIMENT. (M=MODEL NUMBER; RES=RESIDUE NAME; C=CHAIN
REMARK 465 IDENTIFIER; SSSEQ=SEQUENCE NUMBER; I=INSERTION CODE.)
REMARK 465
REMARK 465 M RES C SSSEQI
REMARK 465 CYS B 0
REMARK 465 GLN B 41
REMARK 465 GLY B 42
REMARK 465 VAL B 43
REMARK 465 MET B 44
REMARK 465 VAL B 45
REMARK 465 GLY B 46
REMARK 465 MET B 47
REMARK 465 GLY B 48
REMARK 465 GLN B 49
REMARK 465 LYS B 50
REMARK 465 ASP B 51
REMARK 465 SER M 523
REMARK 465 ASP M 524
In some PDB files, SSBOND records disulfide bond information, while LINK records provide information about additional residue connections. There is no SSBOND information in 4b1y.pdb, and the LINK records describe the coordination bonds of the Mg ion. Because the ion model used here does not use coordination bonds, the LINK section is not kept for this PDB file.
LINK O1G ATP B1377 MG MG B1378 1555 1555 2.09
LINK O1B ATP B1377 MG MG B1378 1555 1555 2.00
LINK MG MG B1378 O HOH B2253 1555 1555 2.09
LINK MG MG B1378 O HOH B2034 1555 1555 2.08
LINK MG MG B1378 O HOH B2273 1555 1555 2.11
LINK MG MG B1378 O HOH B2033 1555 1555 2.07
Use the pdb_filter function to obtain a simplified file. The first two arguments are the input and output file names, respectively. heads specifies the header record types to keep, hetero_residues specifies the non-protein residues to keep, and rename_ions renames ions so that their atom names match the force field.
In Xponge, the default force field names for ions are the fully uppercase element symbols. If the charge is not 1, the charge number is appended. For example, Na+ is NA, Mg2+ is MG2, Fe2+ is FE2, and Fe3+ is FE3.
import Xponge
Xponge.pdb_filter("4b1y.pdb", "4b1y_simple.pdb", heads=["ATOM", "SEQRES", "TER"], hetero_residues=["MG", "ATP", "LAB"], rename_ions={"MG":"MG2"})
3. Building the Force Field
The force fields for the protein and magnesium ions are already available, so directly import the corresponding force fields.
import Xponge.forcefield.amber.ff14sb
import Xponge.forcefield.amber.tip3p
ATP and LAB can use the gaff force field, but their atom types and charges are unknown. We can use the Xponge.Assign structure to specify the force field information. The atom types here use the gaff force field, and the partial charges use the tpacm model. For higher accuracy, the resp charge model can be used.
import Xponge.forcefield.amber.gaff as gaff
assign1 = Xponge.Get_Assignment_From_Mol2("ATP.mol2", total_charge="sum")
assign1.Determine_Atom_Type("gaff")
assign1.Calculate_Charge("tpacm4")
ATP = assign1.toResidueType("ATP")
assign2 = Xponge.Get_Assignment_From_Mol2("LAB.mol2", total_charge="sum")
assign2.Determine_Atom_Type("gaff")
assign2.Calculate_Charge("tpacm4")
LAB = assign2.toResidueType("LAB")
gaff.parmchk2_gaff(ATP, "ATP.frcmod")
gaff.parmchk2_gaff(LAB, "LAB.frcmod")
4. Loading the PDB and Adding Solvent and Ions
C = Xponge.load_pdb("4b1y_simple.pdb", ignore_hydrogen=True, ignore_unknown_name=True, ignore_seqres=False)
C.add_missing_residues()
C.add_missing_atoms()
Xponge.addSolventBox(C, WAT, 25)
Xponge.Solvent_Replace(C, WAT, {K:21+int(round(C.charge)), CL:21})
Xponge.save_pdb(C, "4b1y_final.pdb")
Xponge.save_mol2(C, "4b1y_final.mol2")
Xponge.save_sponge_input(C, "4b1y")
Use VMD to inspect the original 4b1y.pdb and 4b1y_final.mol2. The corresponding loop region can be seen to have been added.
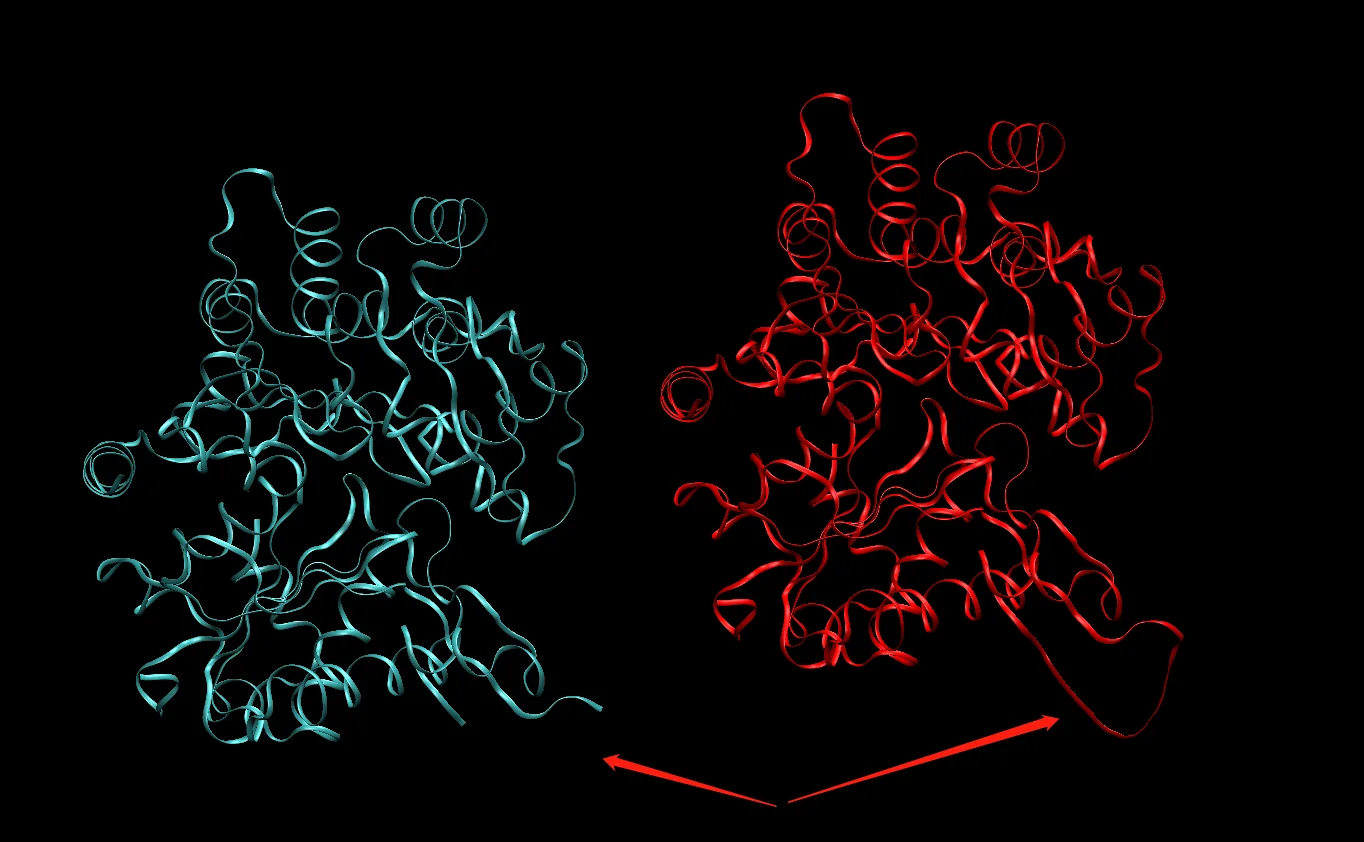
Note that the structure has not been initialized at this point, so the hydrogen positions may not be very good. If a file that does not contain bonding information, such as a PDB file, is used directly for visualization, the displayed structure may be incorrect. This is normal, because MD itself does not directly use PDB as the simulation input.
Use SPONGE to perform minimization.
SPONGE -mode minimization -step_limit 2000 -default_in_file_prefix 4b1y

5. Summary
The Python script used in this tutorial is build.py.
import Xponge
Xponge.pdb_filter("4b1y.pdb", "4b1y_simple.pdb", heads=["ATOM", "SEQRES", "TER"], hetero_residues=["MG", "ATP", "LAB"], rename_ions={"MG":"MG2"})
import Xponge.forcefield.amber.ff14sb
import Xponge.forcefield.amber.tip3p
import Xponge.forcefield.amber.gaff as gaff
assign1 = Xponge.Get_Assignment_From_Mol2("ATP.mol2", total_charge="sum")
assign1.Determine_Atom_Type("gaff")
assign1.Calculate_Charge("tpacm4")
ATP = assign1.toResidueType("ATP")
assign2 = Xponge.Get_Assignment_From_Mol2("LAB.mol2", total_charge="sum")
assign2.Determine_Atom_Type("gaff")
assign2.Calculate_Charge("tpacm4")
LAB = assign2.toResidueType("LAB")
gaff.parmchk2_gaff(ATP, "ATP.frcmod")
gaff.parmchk2_gaff(LAB, "LAB.frcmod")
C = Xponge.load_pdb("4b1y_simple.pdb", ignore_seqres=False)
C.add_missing_residues()
C.add_missing_atoms()
Xponge.addSolventBox(C, WAT, 25)
Xponge.Solvent_Replace(C, WAT, {K:21+int(round(C.charge)), CL:21})
Xponge.save_pdb(C, "4b1y_final.pdb")
Xponge.save_mol2(C, "4b1y_final.mol2")
Xponge.save_sponge_input(C, "4b1y")
Use the following bash commands.
obabel 4b1y_D_ATP.mol2 -O ATP.mol2 -p 7
Xponge name2name -tformat pdb -tfile 4b1y.pdb -tres ATP -fformat mol2 -ffile ATP.mol2 -oformat mol2 -ofile ATP.mol2
obabel 4b1y_C_LAB.mol2 -O LAB.mol2 -p 7
Xponge name2name -tformat pdb -tfile 4b1y.pdb -tres LAB -fformat mol2 -ffile LAB.mol2 -oformat mol2 -ofile LAB.mol2
python build.py