This manual is organized with reference to the GROMACS reference manual [1.1], but it is not a Chinese translation of that manual. Instead, it follows a similar structure to describe the various settings in CudaSPONGE. In fact, after reading the code of many MD simulation packages, one can see that almost every MD package makes appropriate adjustments to algorithms from the literature. CudaSPONGE also contains such adjustments, and explaining them is the purpose of this reference manual.
This manual is incomplete. Because the authors have limited time, and because our highest priority is improving SPONGE, this manual does not aim to be exhaustive. It is still being improved, and some information may be incomplete or not fully correct.
Comments on both form and content are welcome. Relevant information can be added directly in the discussion on this page.
2. SPONGE
“Molecular simulation” is an interesting term. In Chinese, the same phrase can be used to translate two English expressions: molecular simulation and molecular modeling. Although these two expressions can also be translated in ways that distinguish them from each other, in many Chinese contexts both are still rendered as “molecular simulation”. Molecular modeling broadly refers to the general process of describing complex chemical systems through various molecular models, with the goal of understanding and predicting macroscopic properties from detailed atomic-scale knowledge. Molecular simulation, by contrast, usually refers to specific atom-based simulation methods represented by molecular dynamics simulations and Monte Carlo simulations.
The full name of SPONGE is “Simulation Package tOwards Next GEneration molecular modeling”, that is, a simulation package for next-generation molecular modeling. The name contains two uses of “simulation”, but their meanings differ as described above. The name also shows that the head phrase of SPONGE is “Simulation Package”, meaning simulation software. Its core still focuses mainly on molecular simulation, while its goal is to become a next-generation molecular modeling tool, meaning that its direction is more comprehensive and more advanced molecular modeling. At present, SPONGE is mainly a molecular dynamics simulation tool, with several additional molecular simulation capabilities.
3. Basic Mathematical Definitions
3.1 Matrix and Vector Definitions
This document involves many physical quantities. Matrices and vectors are written in bold, such as the force vector F and pressure tensor Π; scalars are written in italics, such as the force magnitude F. A vector is regarded as a matrix whose row or column dimension is 1. A row vector has one row, and a column vector has one column.
Unless otherwise stated, vectors are row vectors. For example, the force vector of a particle has shape (1,3):
F=(FxFyFz)(3-1)
In CudaSPONGE, the corresponding vector is represented by the data structure VECTOR.
For a matrix Y with shape (m,n), its derivative with respect to a scalar x is written as ∂x∂Y. The result is a matrix with shape (m,n), defined as:
For a matrix Y with shape (p,q), its derivative with respect to a matrix X with shape (m,n) is written as ∂X∂Y. The result is a matrix with shape (mn,pq), defined as:
This derivative convention, in which the denominator is expanded first, is called denominator layout. The derivative convention expanded by the numerator is called numerator layout, and the two are transposes of each other.
For a matrix X with shape (m,n) and a matrix Y with shape (n,l), their matrix product is written as XY. The result is a matrix with shape (m,l), defined as:
For a matrix X with shape (m,n) and a matrix Y with shape (m,n), their inner product is written as tr(XTY). The result is a scalar, defined as:
tr(XTY)=i∑mj∑nxijyij(3-5)
For a matrix X with shape (m,n) and a matrix Y with shape (p,q), their outer product is written as X⊗Y. The result is a matrix with shape (mp,qn), defined as:
From the definitions above, several important results follow, including the chain rule for derivatives:
∂X∂Z=∂X∂Y∂Y∂Z(3-7)
For any Y=AXB,
∂X∂Y=B⊗AT(3-8)
3.2 Unit System
CudaSPONGE uses its own unit system internally. Units used for data exchange outside the program are set according to the specific situation.
The internal units are:
Physical quantity
Unit
Explanation
Length
Å
10−10m
Time
20.455 fs
20.455×10−12s
Velocity
Å/(20.455 fs)
Length unit divided by time unit
Charge
18.2223 e
e is the elementary charge
Mass
Da
1/12 of the mass of a carbon-12 atom
Temperature
K
Thermodynamic temperature scale
Energy
kcal/mol
1kcal/mol=4.184kJ/mol
Force
kcal/mol/Å
Pressure
1.4395×10−5bar
NA/(4.184×1028)
The charge unit is chosen so that, in this unit system, the coefficient in Coulomb’s law is 1:
ECoulomb=r12q1q2(3-9)
The time unit is chosen so that, in this unit system, the coefficient in the kinetic-energy calculation is 21:
Ekinetic=21mv2(3-10)
The pressure unit is chosen so that, in this unit system, the coefficient in the calculation of volume work is 1:
W=PV(3-11)
4. Molecular Dynamics
Molecular dynamics is based on classical mechanics. Here it is briefly described through Newtonian mechanics and Hamiltonian mechanics.
4.1 Newtonian Mechanics
For any system, we can calculate the force F at any time, and then use Newton’s laws to calculate particle velocities v and positions r:
{dtdv=mFdtdr=v(4-1)
Integrating this system of differential equations over time t gives the motion of particles over time. In SPONGE, the leapfrog method is used as the integration method:
In Hamiltonian mechanics, the Hamiltonian H is defined using canonical coordinates {q} and canonical momenta {p}. It can usually be written as the sum of kinetic energy T({p}) and potential energy U({q}):
H=T+U(4-3)
Hamilton’s canonical equations are:
{dtdpi=−∂qi∂Hdtdqi=∂pi∂H(4-4)
It is easy to see that equations (4-1) and (4-4) are actually completely equivalent, only written differently.
Phase space is the space containing all canonical coordinates and canonical momenta of the system. At a time t, for an infinitesimal volume element dqdp, the probability density that the system appears at that point is ρ(q,p,t). This quantity is also called the distribution function.
A key equation in molecular dynamics simulation is the Liouville equation, which states that the distribution function is constant along any trajectory in phase space:
dtdρ(q,p,t)=0(4-5)
Expanding equation (4-5) gives:
∂t∂ρ+i∑(∂qi∂ρdtdqi+∂pi∂ρdtdpi)=0(4-6)
Using Hamilton’s canonical equations (4-4) to replace the total time-derivative terms in equation (4-6), and defining the Liouville operator
iL^=i∑(∂pi∂H∂qi∂−∂qi∂H∂pi∂)(4-7)
gives the Liouville equation:
∂t∂ρ=−iL^ρ(4-8)
This equation is very similar to the time-dependent Schrödinger equation, differing only by a factor of ℏ:
iℏ∂t∂φ=H^φ(4-9)
Solving the differential equation shown in equation (4-8) gives:
ρ(q,p,t)=exp(−iL^t)ρ(q,p,0)(4-10)
For this exponential form, there is the Trotter-Suzuki decomposition:
exp(−i(A^+B^)t)≈exp(−iA^t)exp(−iB^t)(4-11)
The Trotter-Suzuki decomposition is very important. First, using t=ΔttΔt, the evolution can be decomposed into (exp(−iL^Δt))Δtt, meaning that a long simulation can be split into many short simulations. Similarly, the kinetic and potential energy terms in H can be separated, meaning that velocities and positions can be updated in sequence. This decomposition is also used later in temperature and pressure control.
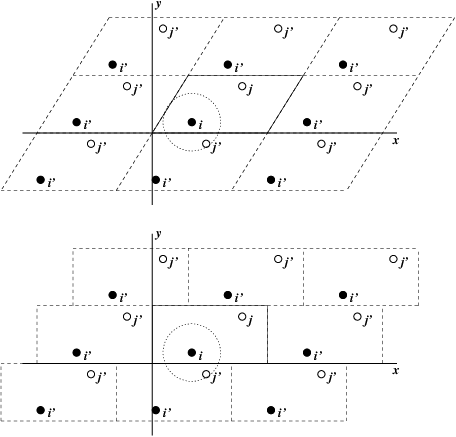
Most simulations use three-dimensional periodic boundary conditions, meaning that the simulation contains only one unit cell, while the system is treated as an infinite spatial repetition of that cell. The following figure is an example of two-dimensional PBC from the GROMACS manual.
A unit cell can be described by the lengths of its three edges, lx, ly, and lz, and the angles α, β, and γ. It can also be described by constructing a box matrix b from the vectors of the three edges. By choosing a suitable coordinate system, the first edge lx can be placed on the x axis, the second edge ly can be placed in the xy plane, and the third edge lz can have an arbitrary position. The box matrix b is then a triangular matrix. Here, each row of the matrix is taken as a row vector of an edge, so the matrix can be written as a lower triangular matrix:
The volume is V=det(b)=bxxbyybzz. The inverse matrix b−1 is also lower triangular, and each of its columns is a column vector in reciprocal space.
The conversion between fractional coordinates s and Cartesian coordinates r for any position or displacement is:
{s=rb−1r=sb(5-2)
Under periodic boundaries, the shortest mapped displacement rij between particles i and j is:
rij=ri−rj−floor((ri−rj)b−1+C)b(5-3)
where C=(0.50.50.5), and floor means taking the floor of each vector component.
In simulations, particle sums are usually required to include only particles within the cutoff distance rc. Particles must not see their own images within rc, so periodic boundaries require the box lengths to be larger than twice the cutoff distance:
⎩⎨⎧lx>2∗rcly>2∗rclz>2∗rc(5-4)
Translating a particle’s Cartesian coordinates by any number of periods is equivalent for the system. It does not affect the simulation itself, only visualization in the output trajectory. When CudaSPONGE stores particle coordinates, it always stores them as positive values closest to the origin while preserving molecular integrity.
5.2 Other Boundary Conditions
The implementation in CudaSPONGE essentially contains only three-dimensional periodic boundary conditions, but by modifying their use they can be treated as other boundary conditions.
For simulations with non-periodic boundary conditions, CudaSPONGE stores an additional set of coordinates without periodic mapping for computing nonbonded interactions. Bonded interactions, however, still use the conditions under periodic boundary conditions, so the box edge lengths must be set large enough in the input to avoid errors.
For pseudo-two-dimensional periodic boundary conditions, the non-periodic dimension can only be the z axis. In the z direction, short-range nonbonded interactions and bonded interactions are still treated as under periodic boundary conditions, so the z-axis box length must be set carefully. For long-range nonbonded interactions, an additional PMC-IZ algorithm is provided [5.1].
6. Constraints
Here, “constrain” and “constraint” are translated as “constraint”, while “restrain” and “restraint” are translated as “restraint”. The distinction is defined as follows: a constraint is enforced by an algorithm, whereas a restraint adds an energy penalty term.
6.1 SETTLE
SETTLE is an iterative analytical solution for preserving the shape of water. By default, if a constraint algorithm is used, SETTLE is applied to water. The SETTLE implementation is based on reference [6.1], but it does not require the triangle to be isosceles and also adds corrections for velocity and pressure.
6.2 SHAKE
SHAKE computes constraints iteratively. Its implementation mainly follows references [6.2] and [6.3].
The main idea is as follows. For a pair of particles whose displacement before the update is r0, after the update the force is repeatedly corrected, and the coordinates and pressure are recalculated, until the distance reaches the preset distance d.
Assuming that the distance after the i-th update is ri, the corrective force F applied in this iteration is:
F=21rir0ri2−d2r0(6-1)
If angles need to be constrained, CudaSPONGE can convert the angle constraint into a distance constraint using the law of cosines. However, such constraints are usually unphysical, difficult to converge, and provide little help for increasing the time step, so constraining angles is not recommended.
7. Temperature Control
Temperature control mainly changes particle velocities. The Trotter-Suzuki decomposition mentioned above tells us that velocity and position updates can be iterated sequentially. Similarly, for temperature control, the corresponding correction to particle velocities can also be iterated separately, although there are several different ways to compute it. We use T^(Δt)=exp(−iL^TΔt) to denote the temperature-control iteration, r^(Δt)=exp(−iL^rΔt) to denote position iteration, and v^(Δt)=exp(−iL^vΔt) to denote velocity iteration. Typical decompositions are:
The first iteration scheme is called the ordinary iteration scheme, and the second is called the middle iteration scheme. The design of the latter mainly follows reference [7.1].
7.1 Andersen Temperature Control
The Andersen thermostat controls temperature by periodically redistributing velocities of particles in an NVE ensemble according to the Maxwell-Boltzmann distribution. In the SPONGE implementation, velocities of all atoms are redistributed after a specified number of steps. The velocity redistribution interval should not be set too small; otherwise, dynamical information is lost and the velocity time autocorrelation function is weakened.
7.2 Berendsen Temperature Control
Berendsen temperature control is a velocity-scaling thermostat. Suppose the system is placed in a heat bath with temperature T0, and the system temperature is T(t). Assume that heat conduction between the system and the heat bath phenomenologically follows Fourier’s law:
dtdT=τ1[T0−T(t)](7-2)
Here, τ has the dimension of time and is therefore called the relaxation time. Taylor-expanding T(t+Δt/2) about t−Δt/2 gives:
The system temperature is controlled by applying this scaling factor to particle velocities.
7.3 Bussi Temperature Control
Bussi temperature control introduces a Wiener random term on the basis of Berendsen temperature control to ensure the correct kinetic-energy distribution:
λ=1+(TT0−T)⋅τΔt+2TfτT0dW(7-6)
Here, f is the number of degrees of freedom of the system, and dW is a Wiener process. Note that in both Berendsen and Bussi temperature control, the scaling factor λ is limited to the range 0.8 to 1.2 to avoid simulation crashes when it becomes too large.
7.4 Langevin Temperature Control
The middle Langevin thermostat introduces fluctuation and damping terms through the Langevin equation to control the system temperature. For particle i, the Langevin equation is:
Here, γ is the friction coefficient, and Ri(t) is a standard Wiener process. The damping force and random force jointly regulate the system temperature and keep it at the desired temperature, with a strict normal distribution. Both Langevin temperature-control algorithms are implemented in SPONGE. The middle Langevin thermostat is superior to the traditional algorithm for sampling the configurational marginal distribution; with the same finite time interval, it can produce more accurate results for configuration-dependent thermodynamic properties.
7.5 Nose-Hoover Chain Temperature Control
8. Pressure Control
Pressure control mainly changes the periodic boundary conditions. Similar to the decomposition in equation (7.1) introduced in the temperature-control section, CudaSPONGE mainly uses the following decomposition for pressure control, where P^(Δt)=exp(−iL^PΔt) denotes the pressure-control iteration:
For the NPT ensemble, the canonical coordinates are now the fractional coordinates si and the components of the box matrix b. We can substitute si and b together into the adiabatic Lagrange equation:
Here, the metric matrix is G=bTb, mg is a generalized mass, and pext is the external pressure. Substitution into the Euler-Lagrange equations gives the dynamical evolution equations for particles and the box:
In the equation above, for the partial derivatives of the potential energy with respect to the box, the independent variables of the potential energy are the box b and the fractional coordinates si. Pext is the external pressure, and γs is the total external surface tension in the xy direction, meaning the sum of the external surface tensions of all surfaces in the xy direction. The second term is the internal pressure. Within the second term, the first subterm is the kinetic contribution to the internal pressure, while the second subterm is the potential-energy contribution, also called the virial. Note that in CudaSPONGE the virial includes the preceding minus sign.
If isotropic, semiisotropic, or semianisotropic changes are needed, constraints can be forcibly imposed on the basis of the anisotropic form. The constrained Π′ values replacing the original Π are:
On this basis, SPONGE also provides an option to forcibly constrain any diagonal element of Π to 0.
If the potential energy U satisfies dU=∑ijdrij∂rij∂U=−∑ijdrijFijT, then the virial calculation can be simplified by expanding the matrices and applying the chain rule:
Similarly, if, when computing some part U0 of the potential energy, it can be guaranteed that the distance between any two particles does not exceed half the box length, then the virial of that part can also be simplified using particle coordinates ri and forces Fi. This is useful for bonded interactions:
Andersen pressure control and Andersen temperature control were both proposed in reference [8.1]. The core idea is to treat the volume as an extended degree of freedom for microcanonical and/or canonical ensemble simulations. It is worth noting that in this system, the canonical coordinates are the fractional coordinates f of atoms rather than their Cartesian coordinates r. Andersen’s original formulation included only isotropic pressure control, while Parrinello and Rahman later proposed an anisotropic pressure-control method based on it (reference [8.2]). In SPONGE, this class of extended-ensemble algorithms is uniformly regarded as an extension of Andersen pressure control. The integration method used here is different from all currently existing methods, but overall it is relatively close to the method of Martyna, Tuckerman, Tobias, and Klein (MTTK pressure control, reference [8.3]). In the pressure-control algorithm proposed by MTTK, the Nose-Hoover algorithm is used to control box velocities. In the SPONGE implementation, the Langevin algorithm is instead used to control box velocities (reference [8.4]). We also take the reciprocal of the number of degrees of freedom to be approximately zero, namely 1/Nf≈0.
Considering only the pressure-control step, the differential expressions for the Cartesian coordinate ri and Cartesian velocity vi of particle i, the Cartesian coordinate of the box b, and the Cartesian velocity of the box g are:
In the isotropic case, Π, R, g, and b in the equation above reduce respectively to the scalar pressure P, random number R, box velocity g, and box length l. The integration formula is then:
Virtual atoms, also called virtual interaction sites, virtual sites, dummy atoms, and so on, are a method for simplifying part of a complex potential energy into another virtual atom. For example, for the potential energy U=r0−6+r0−12, one can define rv=r02. Then U=r0−6+rv−6, which is more symmetric in form, requires only one implementation for computing sixth powers, and may sometimes have clearer physical meaning. Of course, this example is overly simple; without replacement, the potential energy would be even simpler. Common real applications include virtual atoms in TIP4P water molecules. In practice, multi-level virtual atoms can also be defined. For example, for U=r0−6+r0−12+r0−24, one can define r1=r02 and r2=r12, giving U=r0−6+r1−6+r2−6. For convenience below, real atoms are called level-0 virtual atoms. A definition whose highest virtual-atom level is n is called an (n+1)-level virtual atom, and the coordinate of a level-n virtual atom is uniformly denoted by rn.
For simulations containing virtual atoms, virtual atoms are first treated as real atoms in the ordinary calculation. After the calculation, the forces must be redistributed. Differentiating the original form of the potential energy and the form containing virtual atoms gives:
In the equation above, the part outside the parentheses of a partial derivative indicates the quantities held fixed when taking that partial derivative.
According to this equation, force calculation gives:
(∂r0∂U)b=n∑dr0drn(∂rn∂U)ri,i=n,b(9-2)
drn/dr0 can be computed using the chain rule.
For virial calculation, the potential energy must also be expanded in terms of the fractional coordinates fn in the two cases: