高分子建模
SPONGE 1.4 教程。高分子建模
更新时间
2023/04/27
本文以50个链节的聚乙二醇的分子构建(如下图)为例。
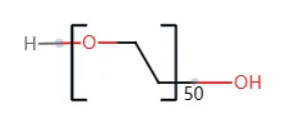
聚乙二醇分子的构建包含下面四种残基:
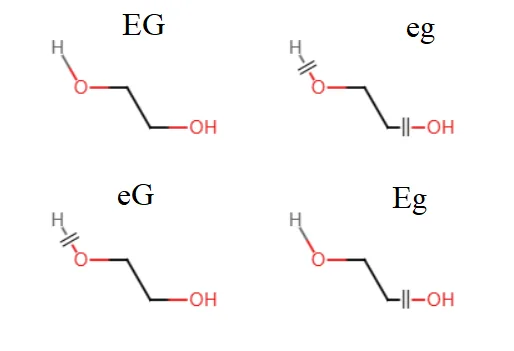
其中,EG、eg、eG、Eg为任意与你现有力场不重复的命名。这里使用的EG是我自己的命名,表示Ethylene glycol的缩写。E表示有头部的氢,e表示无头部的氢;G表示有尾部的羟基,g表示无尾部的羟基。
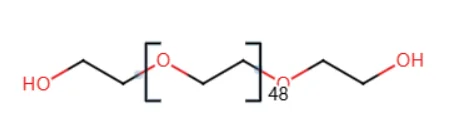
最终目标是构建一个分子
mol = Eg + eg * 48 + eG
也即如下图所示:
1. 构建单体EG力场
import Xponge
import Xponge.forcefield.amber.gaff as gaff
"""从乙二醇的smiles获得assignment,以赋予力场"""
assign = Xponge.Get_Assignment_From_Smiles("OCCO")
"""获得对应的原子类型和电荷"""
assign.determine_atom_type("gaff")
"""如果对精度要求高,使用tpacm4电荷
assign.calculate_charge("resp")
如果对精度要求不高,也可以使用tpacm4电荷"""
assign.calculate_charge("tpacm4")
"""将assignment转为ResidueType"""
EG = assign.to_residuetype("EG")
"""保存文件"""
save_mol2(EG, "EG.mol2")
打开EG.mol2文件可以检查原子类型和电荷等情况
@<TRIPOS>MOLECULE
EG
10 9 1 0 1
SMALL
USER_CHARGES
@<TRIPOS>ATOM
1 O -1.711 0.268 -0.387 oh 1 EG -0.633105
2 C -0.485 -0.387 -0.314 c3 1 EG 0.105505
3 C1 0.603 0.581 0.097 c3 1 EG 0.105505
4 O1 1.843 -0.049 0.177 oh 1 EG -0.633105
5 H -2.186 0.250 0.500 ho 1 EG 0.405197
6 H1 -0.289 -0.876 -1.280 h1 1 EG 0.061202
7 H2 -0.563 -1.156 0.505 h1 1 EG 0.061202
8 H3 0.320 0.955 1.112 h1 1 EG 0.061202
9 H4 0.701 1.428 -0.585 h1 1 EG 0.061202
10 H5 1.767 -1.015 0.175 ho 1 EG 0.405197
@<TRIPOS>BOND
1 1 2 1
2 1 5 1
3 2 3 1
4 2 6 1
5 2 7 1
6 3 4 1
7 3 8 1
8 3 9 1
9 4 10 1
@<TRIPOS>SUBSTRUCTURE
1 EG 1 **** 0 **** ****
从文件中我们可以看见,名为H的氢原子与第一个羟基氧相连,而后面的羟基名字为O1和H5,这两个信息将用于生成其他的残基。
2. 构建残基eg、Eg和eG的力场
"""接着上面构建单体的内容"""
"""构建残基eG"""
eG = EG.deepcopy("eG")
"""Omit_Atoms的第一个参数是要删掉的原子,第二个参数是删掉以后的残基的电荷。
这里删掉了一个氢离子,所以残基电荷应该是-1"""
eG.Omit_Atoms([eG.H], charge=-1)
save_mol2(eG, "EG1.mol2")
"""重复上面过程,构建残基Eg"""
Eg = EG.deepcopy("Eg")
Eg.Omit_Atoms([Eg.O1, Eg.H5], charge=+1)
save_mol2(Eg, "EG2.mol2")
"""重复上面过程,构建残基eg"""
eg = EG.deepcopy("eg")
eg.Omit_Atoms([eg.H, eg.O1, eg.H5], charge=0)
save_mol2(eg, "EG3.mol2")
3. 连接残基
"""接着上面的构建过程"""
"""eg残基的头部原子名是O
eg残基的主链上紧邻头部原子的原子名是C
eg残基的头部原子成键是1.2埃"""
eg.head = "O"
eg.head_next = "C"
eg.head_length = 1.2
"""eg残基的尾部原子名是C1
eg残基的主链上紧邻尾部原子的原子名是C
eg残基的尾部原子成键是1.2埃"""
eg.tail = "C1"
eg.tail_next = "C"
eg.tail_length = 1.2
"""下面设置键角和二面角的连接规则
eg的O、C与其他残基的尾部原子形成109.5度的键角
eg的C、C1其他残基的头部原子与形成109.5度的键角
eg的C1、C、O与其他残基的尾部原子与形成-180度的二面角
eg的O、C、C1与其他残基的头部原子与形成-180度的二面角"""
PI = 3.141592654
eg.head_link_conditions.append({"atoms": ["C", "O"], "parameter": 109.5 / 180 * PI})
eg.tail_link_conditions.append({"atoms": ["C", "C1"], "parameter": 109.5 / 180 * PI})
eg.head_link_conditions.append({"atoms": ["C1", "C", "O"], "parameter": -PI})
eg.tail_link_conditions.append({"atoms": ["O", "C", "C1"], "parameter": -PI})
"""获得分子"""
mol = eg * 50
"""将第一个残基变为Eg
将最后一个残基变为eG
补充上Eg和eG的原子"""
mol.residues[0].set_type("Eg")
mol.residues[-1].set_type("eG")
mol.add_missing_atoms()
"""使用gaff的parmchk2寻找缺失的力场参数"""
gaff.parmchk2_gaff(mol, "eg.frcmod")
"""保存文件"""
save_mol2(mol, "PEG.mol2")
save_sponge_input(mol, "PEG")
4. 观察模型和动力学模拟
使用vmd观察最终生成的PEG.mol2,可见

5. 后记
-
本例只包含聚乙二醇部分的建模,更多的处理如溶剂化等与普通过程相同。
-
例子里的各二面角等可根据自己情况调整。
-
例子中只使用了单体乙二醇进行力场参数的分配,也可以使用聚三乙二醇进行力场参数分配,不同点在于
omit_atoms省略的原子不同 -
如果想构建无限长的分子,可在最后不
set_type,而是对mol连接第0个和最后一个残基的头和尾,也即"""获得分子""" mol = eg * 50 """将第一个残基变为Eg 将最后一个残基变为eG 补充上Eg和eG的原子 mol.residues[0].set_type("Eg") mol.residues[-1].set_type("eG") mol.add_missing_atoms()""" mol.add_residue_link(mol.residues[0].C1, mol.residues[-1].O)