¶ 蛋白质与配体建模
更新时间
2023/05/26
¶ 1. 引言
本文以蛋白序列号4B1Y为例,介绍使用Xponge对包含蛋白质、配体和离子的模型进行构建
使用Xponge版本:1.3.4b3
使用CudaSPONGE版本:1.3
¶ 2. 原始文件的获得与基本文本处理
进入蛋白质数据银行RCSB的4B1Y界面,点击下载对应的PDB和需要用到的小分子配体的mol2文件,如下图所示
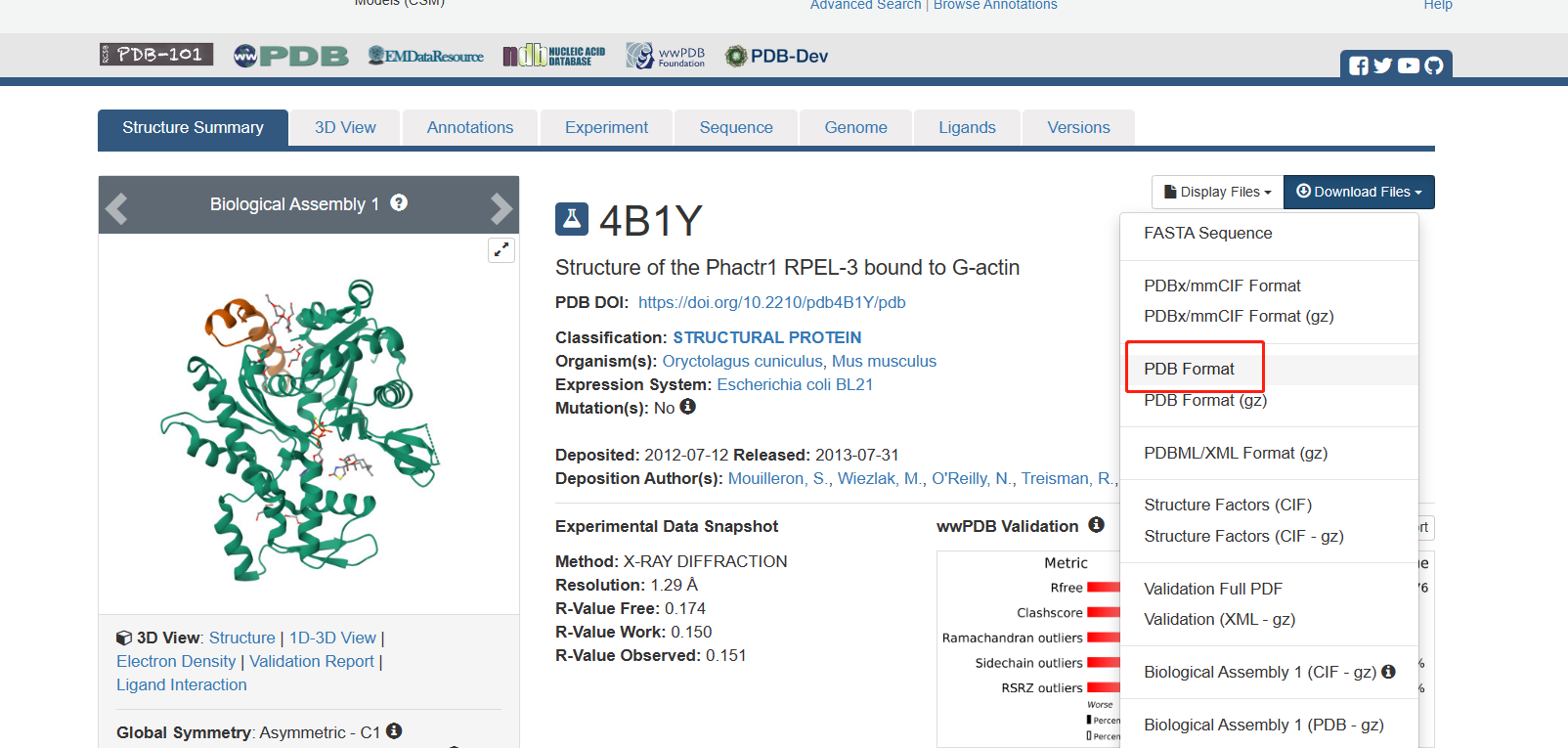
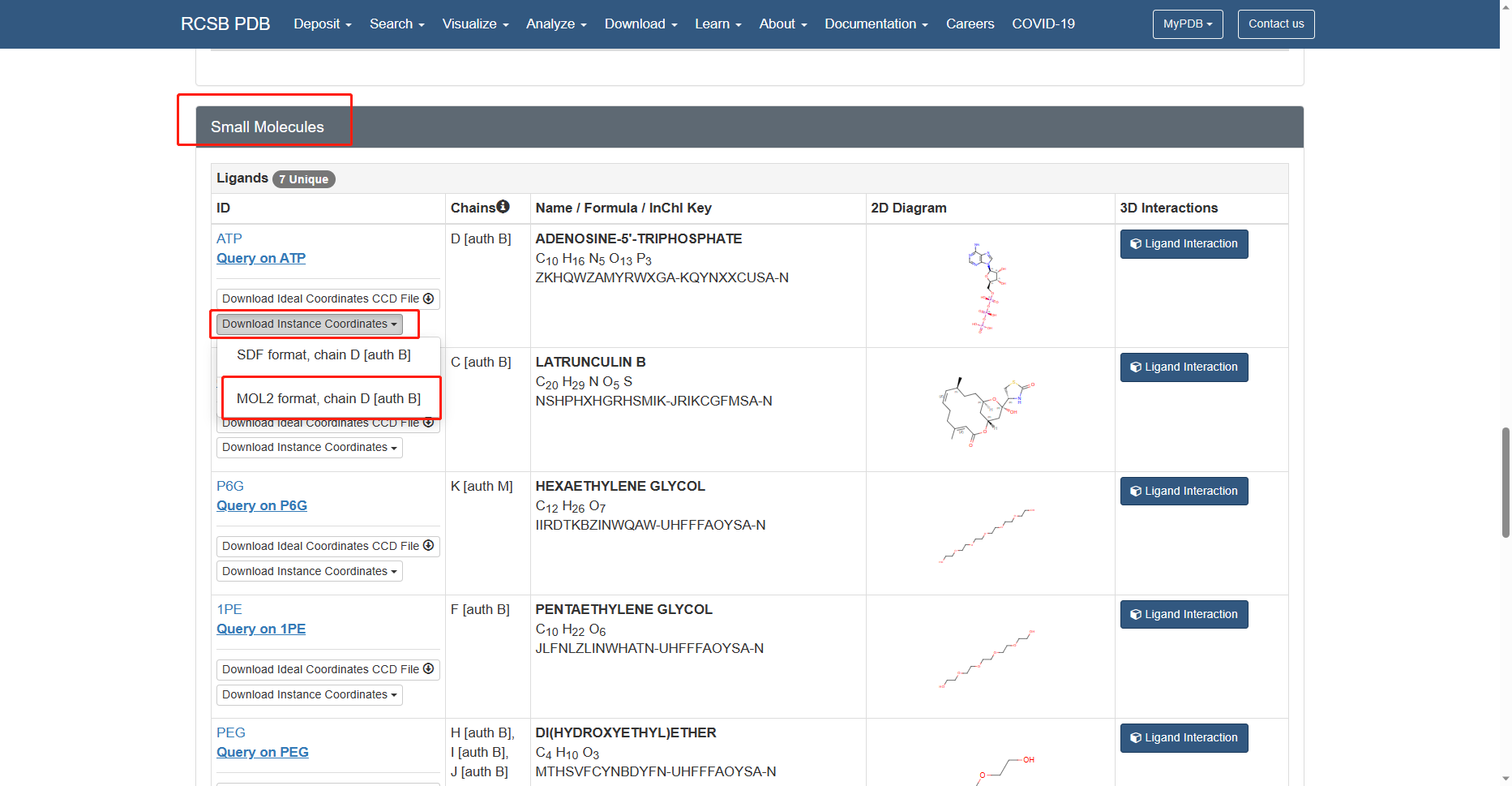
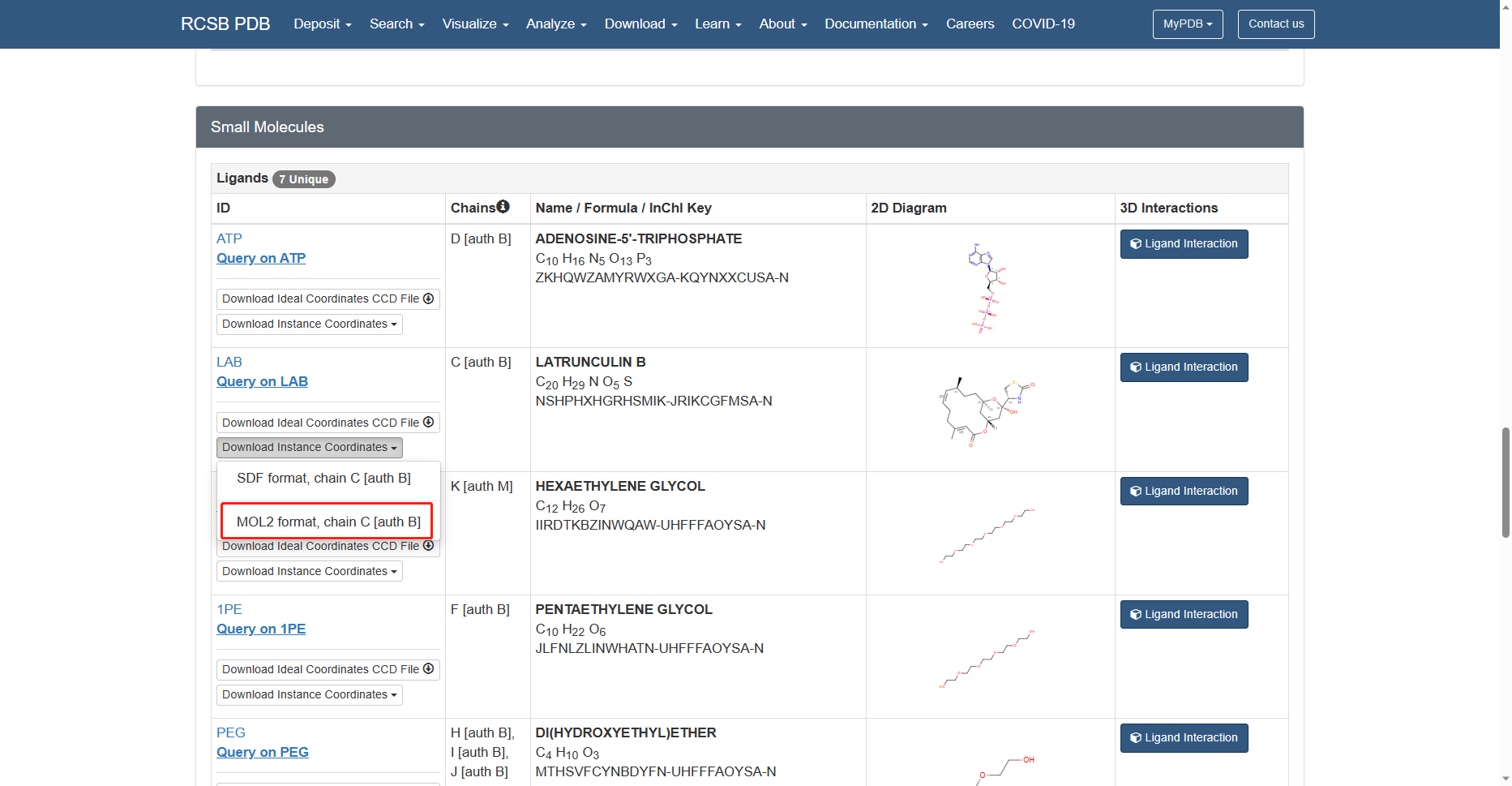
其余的醇和水是蛋白质结晶过程中的小分子,是我们不需要的,因此不下载。
使用openbabel对mol2进行加氢处理,设置pH为7。openbabel加氢后会修改原子名,使用Xponge name2name将原子名修改回pdb中的名字
obabel 4b1y_D_ATP.mol2 -O ATP.mol2 -p 7
Xponge name2name -tformat pdb -tfile 4b1y.pdb -tres ATP -fformat mol2 -ffile ATP.mol2 -oformat mol2 -ofile ATP.mol2
obabel 4b1y_C_LAB.mol2 -O LAB.mol2 -p 7
Xponge name2name -tformat pdb -tfile 4b1y.pdb -tres LAB -fformat mol2 -ffile LAB.mol2 -oformat mol2 -ofile LAB.mol2
虽然pdb文件中也包含小分子的结构信息,但pdb文件中的信息是不完全的,不包含键级及杂化信息,使用openbabel等工具转化时对部分结构的分子可能出错,因此最好是从源头就下载网站中的mol2文件。
打开下载的pdb文件,浏览表头,其中可见REMARK 465部分记述了该pdb中缺少了部分残基,因此需要保留SEQRES部分以完成对缺失残基的补齐
REMARK 465
REMARK 465 MISSING RESIDUES
REMARK 465 THE FOLLOWING RESIDUES WERE NOT LOCATED IN THE
REMARK 465 EXPERIMENT. (M=MODEL NUMBER; RES=RESIDUE NAME; C=CHAIN
REMARK 465 IDENTIFIER; SSSEQ=SEQUENCE NUMBER; I=INSERTION CODE.)
REMARK 465
REMARK 465 M RES C SSSEQI
REMARK 465 CYS B 0
REMARK 465 GLN B 41
REMARK 465 GLY B 42
REMARK 465 VAL B 43
REMARK 465 MET B 44
REMARK 465 VAL B 45
REMARK 465 GLY B 46
REMARK 465 MET B 47
REMARK 465 GLY B 48
REMARK 465 GLN B 49
REMARK 465 LYS B 50
REMARK 465 ASP B 51
REMARK 465 SER M 523
REMARK 465 ASP M 524
在部分PDB中,SSBOND会告诉二硫键信息,而LINK部分会告诉一些额外的残基连接信息。4b1y.pdb中没有SSBOND信息,而LINK信息部分则是Mg离子的配位键。因为用的离子模型不使用配位键,因此该pdb我们也不保留LINK部分。
LINK O1G ATP B1377 MG MG B1378 1555 1555 2.09
LINK O1B ATP B1377 MG MG B1378 1555 1555 2.00
LINK MG MG B1378 O HOH B2253 1555 1555 2.09
LINK MG MG B1378 O HOH B2034 1555 1555 2.08
LINK MG MG B1378 O HOH B2273 1555 1555 2.11
LINK MG MG B1378 O HOH B2033 1555 1555 2.07
我们使用pdb_filter函数来获得简化的文件,其中前两个参数分别是输入和输出的文件名,heads指定需要的表头部分,hetero_residues指定需要的非蛋白残基,rename_ions将离子重命名,以符合力场中原子名。
Xponge里离子的力场名默认是全大写的元素符号,如果电荷不为1则再加上电荷的数字,例如Na+为NA,Mg2+为MG2,Fe2+为FE2,Fe3+为FE3。
import Xponge
Xponge.pdb_filter("4b1y.pdb", "4b1y_simple.pdb", heads=["ATOM", "SEQRES", "TER"], hetero_residues=["MG", "ATP", "LAB"], rename_ions={"MG":"MG2"})
¶ 3. 构建力场
蛋白质和镁离子的力场是已有的,直接import对应力场即可
import Xponge.forcefield.amber.ff14sb
import Xponge.forcefield.amber.tip3p
ATP和LAB可以使用gaff力场,但原子类型和电荷未知,我们可以使用Xponge.Assign结构体来指定力场信息。原子类型这里使用gaff力场,部分电荷使用tpacm模型(更精确可以使用resp电荷模型)
import Xponge.forcefield.amber.gaff as gaff
assign1 = Xponge.Get_Assignment_From_Mol2("ATP.mol2", total_charge="sum")
assign1.Determine_Atom_Type("gaff")
assign1.Calculate_Charge("tpacm4")
ATP = assign1.toResidueType("ATP")
assign2 = Xponge.Get_Assignment_From_Mol2("LAB.mol2", total_charge="sum")
assign2.Determine_Atom_Type("gaff")
assign2.Calculate_Charge("tpacm4")
LAB = assign2.toResidueType("LAB")
gaff.parmchk2_gaff(ATP, "ATP.frcmod")
gaff.parmchk2_gaff(LAB, "LAB.frcmod")
¶ 4. 加载PDB并添加溶剂和离子
C = Xponge.load_pdb("4b1y_simple.pdb", ignore_hydrogen=True, ignore_unknown_name=True, ignore_seqres=False)
C.add_missing_residues()
C.add_missing_atoms()
Xponge.addSolventBox(C, WAT, 25)
Xponge.Solvent_Replace(C, WAT, {K:21+int(round(C.charge)), CL:21})
Xponge.save_pdb(C, "4b1y_final.pdb")
Xponge.save_mol2(C, "4b1y_final.mol2")
Xponge.save_sponge_input(C, "4b1y")
使用VMD观察原始的4b1y.pdb和4b1y_final.mol2,可以看见对应的loop区已经添加上
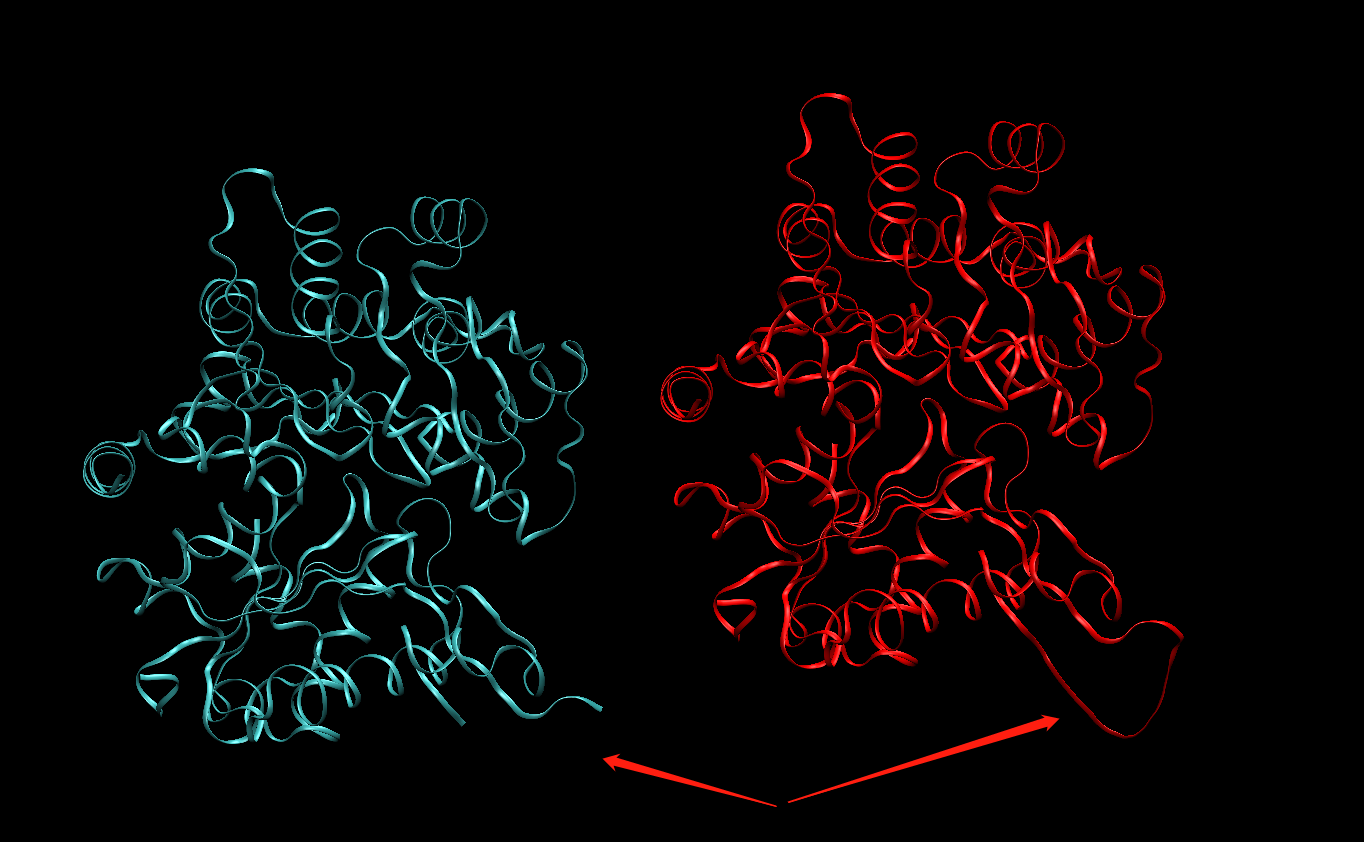
注意,此时的结构未进行初始化,因此氢的位置可能并不太好,直接使用不包含成键信息的文件(如PDB)进行可视化可能得到错误的结构,这是正常的,因为MD本身并不会直接使用PDB作为输入进行模拟。
使用SPONGE做最小化
SPONGE -mode minimization -step_limit 2000 -default_in_file_prefix 4b1y

¶ 5. 总结
本文使用的python脚本build.py
import Xponge
Xponge.pdb_filter("4b1y.pdb", "4b1y_simple.pdb", heads=["ATOM", "SEQRES", "TER"], hetero_residues=["MG", "ATP", "LAB"], rename_ions={"MG":"MG2"})
import Xponge.forcefield.amber.ff14sb
import Xponge.forcefield.amber.tip3p
import Xponge.forcefield.amber.gaff as gaff
assign1 = Xponge.Get_Assignment_From_Mol2("ATP.mol2", total_charge="sum")
assign1.Determine_Atom_Type("gaff")
assign1.Calculate_Charge("tpacm4")
ATP = assign1.toResidueType("ATP")
assign2 = Xponge.Get_Assignment_From_Mol2("LAB.mol2", total_charge="sum")
assign2.Determine_Atom_Type("gaff")
assign2.Calculate_Charge("tpacm4")
LAB = assign2.toResidueType("LAB")
gaff.parmchk2_gaff(ATP, "ATP.frcmod")
gaff.parmchk2_gaff(LAB, "LAB.frcmod")
C = Xponge.load_pdb("4b1y_simple.pdb", ignore_seqres=False)
C.add_missing_residues()
C.add_missing_atoms()
Xponge.addSolventBox(C, WAT, 25)
Xponge.Solvent_Replace(C, WAT, {K:21+int(round(C.charge)), CL:21})
Xponge.save_pdb(C, "4b1y_final.pdb")
Xponge.save_mol2(C, "4b1y_final.mol2")
Xponge.save_sponge_input(C, "4b1y")
使用以下bash命令
obabel 4b1y_D_ATP.mol2 -O ATP.mol2 -p 7
Xponge name2name -tformat pdb -tfile 4b1y.pdb -tres ATP -fformat mol2 -ffile ATP.mol2 -oformat mol2 -ofile ATP.mol2
obabel 4b1y_C_LAB.mol2 -O LAB.mol2 -p 7
Xponge name2name -tformat pdb -tfile 4b1y.pdb -tres LAB -fformat mol2 -ffile LAB.mol2 -oformat mol2 -ofile LAB.mol2
python build.py